Data Inputs¶
Reading in RNA Sequence Data¶
-
read_rna_file(rna_file_path, identifier='hugo', fetch_missing_hugo=False)¶ - Read in a cbioportal pancancer or TCGA Xena Hub txt file containing mixture gene expression data, and return a Pandas DataFrame. Processing includes:
- removing genes with no identifier
- removing duplicate genes (keep the first occurance)
- removing genes with NaN values
Parameters: - file_path – String. Relative or full path to the RNA Seq file. This file is tab seperated and includes two columns, ‘Hugo_Symbol’ and ‘Entrez_Gene_Id’, for each gene preceding the expression values for each patient
- identifier – String. Determines which gene identifier to use to index the rna data. Must be set to either: - ‘hugo’ for Hugo Symbols - ‘entrez’ for Entrez Gene ID
- fetch_missing_hugo – Boolean. Whether to fetch missing Hugo Symbols (by Entrez Gene ID) from ncbi website
Returns: Pandas DataFrame, indexed by either Hugo_Symbol or Entrez_Gene_Id, containing RNA expression data for each patient. Rows are genes, columns are patients.
In addition to reading in an already-downloaded RNA dataset, you can fetch datasets directly from either cBioPortal or UCSC Xena directly:
-
download_by_name(source, type, download_to=td.get_td_Home()+"data/downloaded/", fetch_missing_hugo=False): Downloads TCGA RNA Sequence data from cBioPortal and UCSC Xena Hub.
Parameters: - source – String. Must be either “xena” or “cbio”. Where to download data from.
- type – String. Must match the specific identifier used for a cancer type by the given source
- download_to – String. Full or relative path to directory to save downloaded data to. Default is the data/downloads folder within the package’s library folder (OS/install dependent)
- fetch_missing_hugo – Boolean. Whether to fetch missing Hugo Symbols (by Entrez Gene ID) from ncbi website
Returns: Pandas DataFrame, indexed by either Hugo_Symbol or Entrez_Gene_Id, containing RNA expression data for each patient. Rows are genes, columns are patients.
The current list of valid strings for type, as of March 2022, is the following:
cBioPortal: ‘Acute Myeloid Leukemia’, ‘Adrenocortical Carcinoma’, ‘Bladder Urothelial Carcinoma’, ‘Brain Lower Grade Glioma’, ‘Breast Invasive Carcinoma’, ‘Cervical Squamous Cell Carcinoma’, ‘Cholangiocarcinoma’, ‘Colorectal Adenocarcinoma’, ‘Diffuse Large B-Cell Lymphoma’, ‘Esophageal Adenocarcinoma’, ‘Glioblastoma Multiforme’, ‘Head and Neck Squamous Cell Carcinoma’, ‘Kidney Chromophobe’, ‘Kidney Renal Clear Cell Carcinoma’, ‘Kidney Renal Papillary Cell Carcinoma’, ‘Liver Hepatocellular Carcinoma’, ‘Lung Adenocarcinoma’, ‘Lung Squamous Cell Carcinoma’, ‘Mesothelioma’, ‘Ovarian Serous Cystadenocarcinoma’, ‘Pancreatic Adenocarcinoma’, ‘Pheochromocytoma and Paraganglioma’, ‘Prostate Adenocarcinoma’, ‘Sarcoma’, ‘Skin Cutaneous Melanoma’, ‘Stomach Adenocarcinoma’, ‘Testicular Germ Cell Tumors’, ‘Thymoma’, ‘Thyroid Carcinoma’, ‘Uterine Carcinosarcoma’, ‘Uterine Corpus Endometrial Carcinoma’, ‘Uveal Melanoma’
UCSC Xena: ‘Acute Myeloid Leukemia’, ‘Adrenocortical Cancer’, ‘Bile Duct Cancer’, ‘Bladder Cancer’, ‘Breast Cancer’, ‘Cervical Cancer’, ‘Colon and Rectal Cancer’, ‘Colon Cancer’, ‘Endometrioid Cancer’, ‘Esophageal Cancer’, ‘Glioblastoma’, ‘Head and Neck Cancer’, ‘Kidney Chromophobe’, ‘Kidney Clear Cell Carcinoma’, ‘Kidney Papillary Cell Carcinoma’, ‘Large B-cell Lymphoma’, ‘Liver Cancer’, ‘Lower Grade Glioma’, ‘Lower Grade Glioma and Glioblastoma’, ‘Lung Adenocarcinoma’, ‘Lung Cancer’, ‘Lung Squamous Cell Carcinoma’, ‘Melanoma’, ‘Mesothelioma’, ‘Ocular Melanomas’, ‘Ovarian Cancer’, ‘Pancreatic Cancer’, ‘Pheochromocytoma and Paraganglioma’, ‘Prostate Cancer’, ‘Rectal Cancer’, ‘Sarcoma’, ‘Stomach Cancer’, ‘Testicular Cancer’, ‘Thymoma’, ‘Thyroid Cancer’, ‘Uterine Carcinosarcoma’
Examples:
>>> import os
>>> rna_cbio = td.download_by_name('cbio', 'Ovarian Serous Cystadenocarcinoma', download_to=os.getcwd(), fetch_missing_hugo=True)
Downloading data from cbioportal to /Users/<username>/ov_tcga_pan_can_atlas_2018.tar.gz...
100% [......................................................................] 103274157 / 103274157
Fetching missing Hugo Symbols for genes by Entrez ID...
Found 1 / 13 of the missing values
>>> print(rna_cbio)
TCGA-04-1348-01 TCGA-04-1357-01 TCGA-04-1362-01 ... TCGA-OY-A56Q-01 TCGA-VG-A8LO-01 TCGA-WR-A838-01
Hugo_Symbol ...
UBE2Q2P2 25.716221 20.473921 29.909980 ... 15.716029 28.912917 59.400754
HMGB1P1 321.241331 128.865135 424.021806 ... 400.614342 309.174887 431.623999
LOC155060 222.150613 244.412320 377.449014 ... 387.776781 412.614342 540.597847
RNU12-2P 0.676632 0.090280 0.451162 ... 0.453561 7.464714 4.282079
EZHIP -0.059851 -0.479632 4.514755 ... -0.306245 0.693589 1.320945
... ... ... ... ... ... ... ...
ZYG11A 6.824650 4.775693 66.800880 ... 141.004912 7.760542 79.382924
ZYG11B 483.942790 518.654716 791.336454 ... 1000.073852 1321.299392 1068.161532
ZYX 9692.870954 3284.754545 3747.937644 ... 6698.448007 4961.305257 3907.580348
ZZEF1 429.818654 684.474498 766.249800 ... 448.557587 709.015420 1665.424483
ZZZ3 475.233973 414.753874 802.717598 ... 2462.888278 1295.464299 1443.856175
[19064 rows x 300 columns]
>>> rna_xena = td.download_by_name('xena', 'Ovarian Cancer', fetch_missing_hugo=False)
Downloading data from Xena Hub to /opt/py3.9env/lib/python3.9/site-packages/TumorDecon/data/downloaded//HiSeqV2.gz...
100% [........................................................................] 16904473 / 16904473
>>> print(rna_xena)
TCGA-61-1910-01 TCGA-61-1728-01 TCGA-09-1666-01 ... TCGA-29-1702-01 TCGA-24-1417-01 TCGA-57-1585-01
Hugo_Symbol ...
ARHGEF10L 775.638814 638.278161 292.069698 ... 285.819639 388.964774 1091.183840
HIF3A 8.704138 163.905991 50.190351 ... 1966.434649 522.229474 510.680697
RNF17 0.000000 0.000000 0.000000 ... 0.000000 0.000000 0.000000
RNF10 5536.810611 3688.475448 2081.065875 ... 2204.056642 3539.408216 3186.036883
RNF11 1098.628128 1764.914771 2540.034063 ... 2478.786156 2102.389042 1811.543621
... ... ... ... ... ... ... ...
PTRF 2672.103531 1843.865596 3230.078307 ... 2975.497567 4477.500339 8065.348376
BCL6B 105.418119 36.424211 41.130701 ... 28.153557 140.337452 245.065741
GSTK1 5985.512182 5203.702549 6308.893763 ... 1089.444025 4591.614580 2086.123131
SELP 1.934267 15.729240 4.905667 ... 2.928014 57.688259 105.403367
SELS 1351.019271 691.226326 645.676332 ... 1017.549232 503.321528 551.526558
[20530 rows x 308 columns]
Reading in Signature Matrices and Gene Sets¶
The examples in this User Guide use mostly the LM22 signature matrix, or in the case of rank-based methods, an up-regulated gene set derived from LM22. These cell signatures are included in the package. However, TumorDecon can also be run with any user-provided cell signatures.
-
read_sig_file(file_path, delim="\t", geneID="Hugo_Symbol")¶ Read in a signature matrix (signature gene expression data for a number of different cell types) from a .csv or .txt file, and (if applicable) convert the gene identifiers from Entrez/Ensembl Gene ID to Hugo Symbols. Returns a Pandas DataFrame, indexed by ‘Hugo_Symbol’, where the rows are the genes and the columns are the cell types you wish to include. If no
file_pathprovided, this function returns the LM22 signature matrix.Parameters: - file_path – String. Relative or full path to signature matrix file. The first row of the file should include the names of your cell types, and the first column should be either Hugo Symbols or Entrez Gene IDs. If no file_path given, LM22 is assumed.
- delim – String. Delimiter to use. Default is ‘\t’ (tab separated)
- geneID – String. Must be either “Hugo_Symbol”, “Ensembl_Gene_ID”, “Entrez_Gene_ID”. Describes how genes are labeled in the signature matrix file
Returns: Pandas DataFrame containing signature expression values of each gene for each cell type. Columns are the cell types, rows are the genes (indexed by Hugo Symbol).
Example: Assume we have a tab-separated file called “my_sig_matrix.txt”, where the genes are reference by Ensembl ID:
gene CD8_subtype_1 CD8_subtype_2 CD4_subtype_1 CD4_subtype_2 B_cell_subtype_1 B_cell_subtype_2 NK_subtype_1 NK_subtype_2 mono_subtype_1 mono_subtype_2 Endothelial_subtype_1 Endothelial_subtype_2 Fibroblast_subtype_1 Fibroblast_subtype_2 Neutrophils_subtype_1 Neutrophils_subtype_2
ENSG00000000938 116.0001394 24.38016936 8.453628541 1.672547127 36.5573964 73.34069196 407.2145862 156.8132734 447.4100013 557.9327456 24.76138974 2.174048414 2.142543208 5.412057344 558.770835 739.358363
ENSG00000001167 22.50340273 0.75239339 15.05620142 12.07992823 10.26260586 8.947390096 10.62795065 12.4308104 9.912563566 13.39346071 7.472100959 22.07936334 7.783380993 2.073507549 46.75973 37.21335
ENSG00000002834 154.161176 36.456214 110.4616367 99.4175757 59.25913937 51.64127258 104.0716383 191.6722237 76.53034102 94.86365533 65.90175897 15.73233353 28.93766895 10.79387212 259.2520035 250.373201
... ....... .......
We can read this into python with:
>>> sig = td.read_sig_file("my_sig_matrix.txt", geneID='Ensembl_Gene_ID')
>>> print(sig.head())
CD8_subtype_1 CD8_subtype_2 CD4_subtype_1 ... Fibroblast_subtype_2 Neutrophils_subtype_1 Neutrophils_subtype_2
Hugo_Symbol ...
FGR 116.000139 24.380169 8.453629 ... 5.412057 558.770835 739.358363
NFYA 22.503403 0.752393 15.056201 ... 2.073508 46.759730 37.213350
LASP1 154.161176 36.456214 110.461637 ... 10.793872 259.252004 250.373201
TSR3 39.389183 48.998150 16.131921 ... 0.371443 2.038300 3.715013
NADK 16.429699 12.887361 9.760582 ... 1.503814 871.354356 1006.496017
[5 rows x 16 columns]
-
read_geneset(file_path)¶ Read in a .csv file containing the up / down regulated genes for each cell type
Parameters: file_path – String. Relative or full path to csv file. File should have columns named for each cell type, and rows must contain Hugo Symbols for genes to be considered as up (or down) regulated for that cell type. If not all cell types have the same number of up(down) regulated genes, excess rows in each column should be coded as “NaN” - If no file_path given, the up-regulated gene set derived in Le et al. (2020) is assumed. Returns: Dictionary. Keys are the column names of the given csv file (cell types), values are a list of up (or down) regulated genes for that cell type
Creating Custom Lists of Up/Down-Regulated Genes from Signature Matrix¶
The following function provides a method for determining a list of up-regulated and down-regulated genes from a given signature matrix:
-
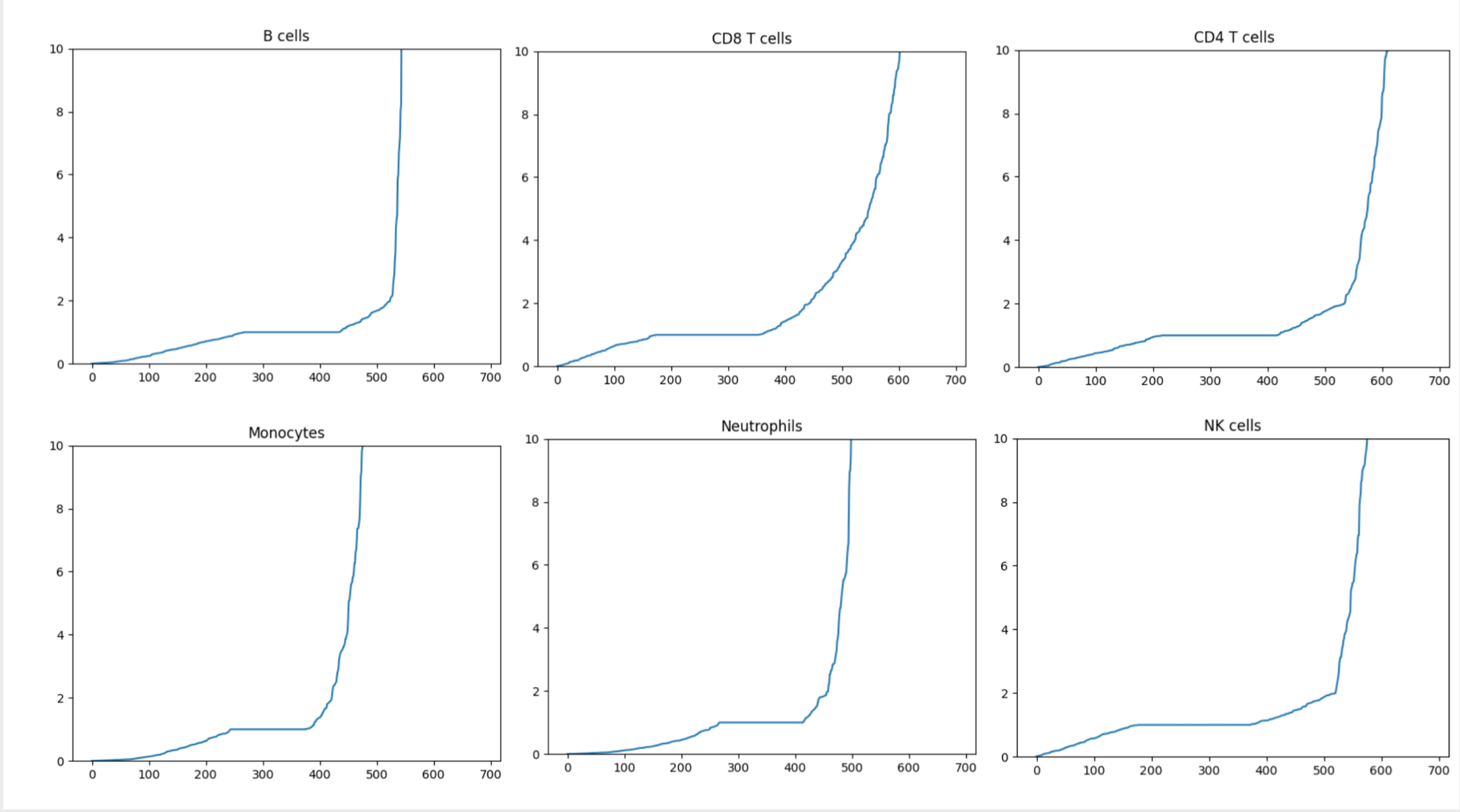
find_up_down_genes_from_sig(sig_df, down_cutoff=0.4, up_cutoff=4.0, show_plots=False)¶ Given a signature matrix,
`sig_df, find a list of up-regulated and down-regulated genes for each cell type in the signature, using the following method:- Divide each gene expression value in Signature Matrix by the median value of the given gene across all cell types.
- All genes with ratios below
down_cutoffare considered “down-regulated genes” while all genes with ratios aboveup_cutoffare considered “up-regulated genes”.
Parameters: - sig_df – Pandas DataFrame. Signature matrix (rows are genes, columns are cell types) indexed by Hugo Symbol
- down_cutoff – Float. Value to use for the cutoff point as to what should be considered a “down-regulated gene”
- up_cutoff – Float. Value to use for the cutoff point as to what should be considered an “up-regulated gene”
- show_plots – Boolean. Whether to show a plot of the sorted signature matrix ratios for each cell type (Can be used to help choose the cutoff points).
Return up: Dictionary. Keys are cell types, values are a List of up-regulated genes for that cell type
Return down: Dictionary. Keys are cell types, values are a List of down-regulated genes for that cell type
Example:
>>> LM6 = td.read_sig_file('LM6.txt')
>>> up_geneset_LM6, down_geneset_LM6 = td.find_up_down_genes_from_sig(LM6, down_cutoff=0.4, up_cutoff=4.0, show_plots=True)